La Integritat de les dades És un pilar central en la indústria farmacèutica i en qualsevol entorn regulat per GxP. Tant el FDA (Estats Units), com el EMA (Europa) i la MHRA (Regne Unit) han desenvolupat guies específiques que aborden els requisits per garantir que les dades generades al llarg del cicle de vida del producte siguin complet, coherent, precís, disponible i atribuïble.
Tot i que es comparteixen els principis fonamentals, cada autoritat introdueix matisos importants que determinen la manera com les empreses han d'implementar els seus sistemes de control, automatització i garantia de qualitat.
Aquest article ofereix un comparació pràctica per facilitar a les organitzacions l'aplicació de controls sòlids i defensables per auditoria.
1. Principis fonamentals compartits (FDA, EMA, MHRA)
Tot i que cada agència reguladora ofereix els seus propis matisos, Tots tres coincideixen en un conjunt de principis essencials que formen la base de qualsevol sistema sòlid d'integritat de dades. Aquests principis s'apliquen a tots els processos GxP i van des de la captura i el processament de dades inicials fins a l'arxiu, l'accés, la revisió i la recuperació al llarg del cicle de vida.
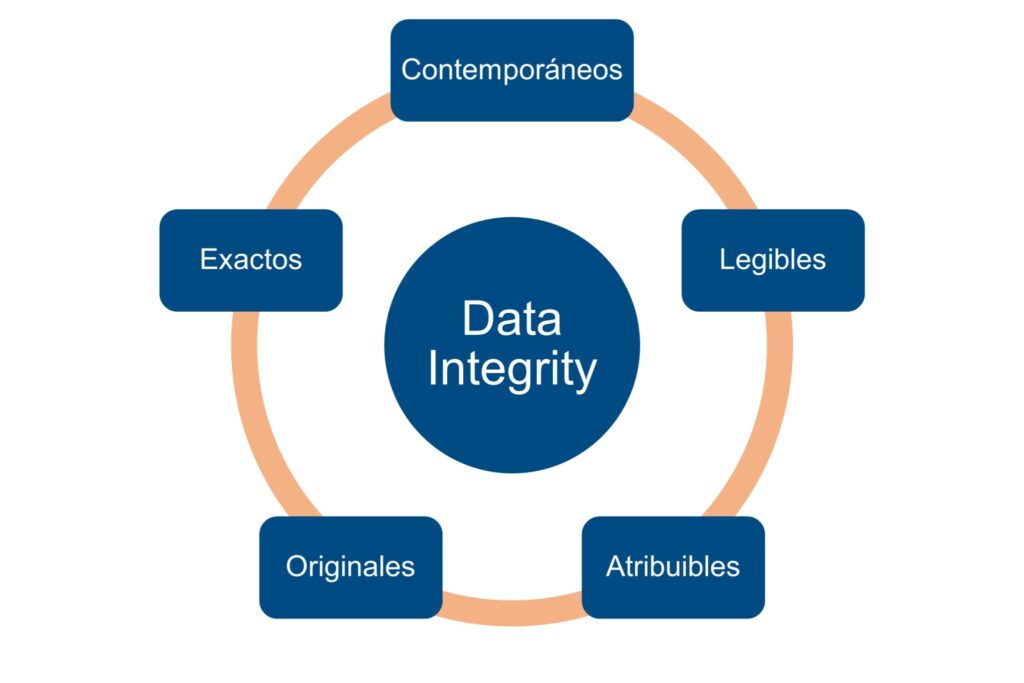
1.1. Integritat de dades suportada per ALCOA+
Les tres autoritats reguladores consideren ALCOA+ com l'estàndard conceptual per definir què són dades fiables en contextos GxP. Les dades han de ser:
- Atribuïble: clarament associat a una persona, equip o sistema.
- Llegible: comprensible durant tot el cicle de vida.
- Contemporanis: registrats en el moment en què es produeixen.
- Originals: conservats en la seva forma primària o amb còpies fidels.
- Exacte: lliure de manipulacions i errors.
I, a més: Completa, consistent, Durable y Disponible.
L'evolució normativa impulsada per la digitalització ha portat fins i tot a l'adopció del terme ALCOA++ en documents recents de l'EMA, especialment en les directrius per a sistemes informàtics i dades electròniques en assaigs clínics (GCP). Aquesta expansió pretén incorporar requisits de traçabilitat i governança de dades encara més avançats per gestionar sistemes informàtics sofisticats i l'ús de tecnologies híbrides.
Aquest marc conceptual és la base de cada inspecció de la FDA, EMA i MHRA, que ara avaluen no només les dades en si, sinó també la traçabilitat lògica entre registres, decisions i proves.
1.2. Cicle de vida de les dades: enfocament holístic i continu
Les tres agències entenen la integritat de les dades com un procés transversal que va més enllà de la mera adquisició:
- Generació
- Acusació
- Revisió
- Informeu
- Arxivat
- Recuperació
- Modificació (controlada)
- Retirada o destrucció
La FDA, l'EMA i la MHRA esperen que cada etapa tingui controls tècnics i procedimentals, i que l'organització pugui demostrar la seva robustesa en les auditories.
1.3. Enfocament basat en el risc
La governança de les dades apareix explícitament a MHRA i transversalment a la FDA i l'EMA. Tots estan d'acord que el La integritat de les dades no pot dependre únicament de les eines tecnològiques. Hi ha d'haver:
- Cultura de la transparència.
- Clar els rols i les responsabilitats.
- Tolerància zero a manipulacions o retards en el registre.
- Disponibilitat de recursos suficients en QA.
- Autonomia de l'àrea de qualitat per revisar, bloquejar o investigar registres.
Aquest enfocament cultural és ara un criteri clau en les inspeccions: els reguladors busquen demostrar si l'empresa "viu" la integritat de les dades o simplement la documenta.
1.4. Enfocament basat en el risc segons ICH Q9
La La gestió del risc regula com s'han d'aplicar els controls. La FDA, l'EMA i la MHRA esperen:
- Identificació de vulnerabilitats de dades.
- Classificació del risc segons impacte en el producte, pacient i integritat.
- Mitigació alineada amb GAMP 5 (2a Ed.).
- Revisió contínua del risc després de canvis de programari, processos o personal.
En les auditories, els inspectors ja no només pregunten sobre els SOP: ara demanen proves que s'han identificat, avaluat i mitigat activament els riscos de les dades.
1.5. Controls tècnics sòlids: pista d'auditoria, permisos i traçabilitat
Les tres agències estan d'acord que els sistemes informàtics ho han de fer integrar controls que eviten la manipulació:
- Pista d'auditoria segura i inalterable, registrant qui va fer què, quan i per què.
- Gestió d'accés basada en rols, sense comptes genèrics ni permisos innecessaris.
- Control de versions, tant de programari com de configuracions crítiques.
- Còpia de seguretat i recuperació de dades, validades i provades periòdicament.
- Sincronització horària, essencial per a la correlació d'esdeveniments.
La diferència rau en el grau d'exigència (MHRA és el més estricte), però el principi és idèntic.
1.6. Revisió periòdica de la pista d'auditoria i dades crítiques
És un dels elements de major atenció en les inspeccions durant els darrers anys. FDA, EMA i MHRA requereixen:
- Revisió rutinària de la pista d'auditoria basada en el risc.
- Evidència que les revisions són independents, oportunes i competents.
- Formació específica del personal revisor.
- Coherència entre el registre electrònic i les operacions reals.
- No n'hi ha prou amb "activar" la pista d'auditoria: ha de ser útil, revisada i defensable.
1.7. Validació del sistema informàtic (CSV/CSA) com a part bàsica del control
Els reguladors accepten que un sistema no validat és un sistema que no garanteix la integritat de les dades. Expectatives comunes:
- Validació proporcional al risc (GAMP 5 / CSA).
- Traçabilitat completa URS → Riscos → Proves → Evidències.
- Proves significatives i no redundants.
- Gestió del cicle de vida del sistema: canvis, incidències, manteniment.
La FDA ha impulsat la visió de la CSA, però l'EMA i la MHRA estan d'acord en el seu enfocament basat en el risc.
2. Diferències entre FDA, EMA i MHRA
A continuació es mostren les principals diferències entre la FDA, l'EMA i la MHRA pel que fa a l'enfocament i la terminologia, el grau d'independència de la pista d'auditoria, les expectatives dels sistemes híbrids i la revisió de dades i la segregació de responsabilitats:
Enfocament i terminologia
| FDA | Adopta un to més operatiu, centrant-se en els casos reals observats durant les inspeccions. El seu enfocament és pragmàtic des del punt de vista del compliment. Publicació clau: "Integritat de les dades i compliment de les preguntes i respostes del CGMP de drogues". |
| EMA | Integra la integritat de les dades dins del marc europeu GMP, enllaçant la integritat de les dades amb el sistema de qualitat farmacèutica (ICH Q10). S'orienta cap a les expectatives estratègiques: govern del sistema, rols i responsabilitats. |
| MHRA | És l'autoritat històricament més estricta. La seva guia va més enllà de GMP i introdueix conceptes com el "sistema de govern de dades" i un alt nivell de maduresa en els controls. |
Grau d'independència de la pista d'auditoria
| FDA | Subratlla la necessitat que la pista d'auditoria sigui segura i no editable, però no requereix una arquitectura específica. |
| EMA | Insistir que la pista d'auditoria es revisi de manera rutinària com a part del flux de treball de qualitat. |
| MHRA | És clar: les pistes d'auditoria no es poden desactivar, han d'estar totalment integrades i protegides d'accés administratiu inadequat. |
Expectatives sobre els sistemes híbrids
| FDA | Els accepta, però adverteix dels seus riscos: duplicació de fonts, retards, manipulacions i errors manuals. |
| EMA | Afavoreix la migració progressiva cap a sistemes integrats. |
| MHRA | És més contundent: els sistemes híbrids s'han de considerar una solució temporal, no permanent. |
Revisió de dades i segregació de responsabilitats
| FDA | Els accepta, però adverteix dels seus riscos: duplicació de fonts, retards, manipulacions i errors manuals. |
| EMA | Afavoreix la migració progressiva cap a sistemes integrats. |
| MHRA | És més contundent: els sistemes híbrids s'han de considerar una solució temporal, no permanent. |
Comparació pràctica
|
SEGURETAT DE SISTEMES INFORMÀTICS |
|||
|
Requisit |
FDA | EMA |
MHRA |
|
Gestió d'accés |
Necessari, basat en rols | Integrat en PQS | Molt estricte i detallat |
|
Pista d'auditoria |
Obligatori i revisable | Obligatori amb revisió periòdica | No es pot desactivar |
|
Control de versions |
Recomanat | Sol·licitat | Obligatori i supervisat |
|
REGISTRES EN PAPER I SISTEMES HÍBRIDS |
|||
|
Aspecte |
FDA | EMA |
MHRA |
|
Acceptació |
Limitat, amb riscos | Acceptat però no preferit | Transitòria, no recomanable |
|
Principals riscos |
Errors, duplicitat | Manca de traçabilitat | Manipulació i pèrdua de control |
|
GOVERNAMENT DE DADES |
|||
| Element | FDA | EMA |
MHRA |
|
Model formal de “govern de dades”. |
No explícit | Integrat a PQS | Requisit explícit i detallat |
3. Com implementar un sistema robust que compleixi amb la FDA, EMA i MHRA
La implementació d'un sistema d'integritat de dades realment robust implica anar més enllà del compliment documental i avançar cap a a Marc integral que combina tecnologia, cultura organitzativa, govern clar i supervisió contínua. Tot i que cada agència té particularitats, tots coincideixen que Data Integrity ho ha de fer gestionats al llarg de tot el cicle de vida, des del disseny del sistema fins a la jubilació. Això vol dir que els requisits no es poden abordar de manera reactiva; S'han d'integrar des de les primeres etapes per garantir que el sistema sigui inherentment fiable, defensable i capaç de suportar auditories rigoroses.
Un primer pas crític és adoptar un enfocament de "Integritat de les dades per disseny". Aquest principi, cada cop més present a les directrius de la FDA i l'EMA, passa per configurar sistemes digitalitzats i equips GxP amb capes de control tècnic dissenyades des de la seva concepció. La definició correcta dels requisits d'usuari (URS) és essencial per garantir que el funcionalitats principals s'incorporen abans de la validació, com ara la traçabilitat, la pista d'auditoria inalterable, la gestió d'accés o els controls de versions. Un sistema que no pot registrar dades de manera segura o que no permet restringir adequadament els permisos dels usuaris serà, per definició, un sistema incompatible, per més que estigui complementat amb procediments.
La validació del sistema informatitzat (CSV), alineada amb les expectatives GAMP 5 i FDA CSA, és un altre pilar fonamental. La validació ha de demostrar que el sistema funciona tal com està dissenyat, que protegeix les dades mitjançant controls tècnics sòlids i que funciona en un estat controlat en condicions rutinàries. Aquest procés ha d'incloure proves específiques que garanteixin la integritat dels registres electrònics, la fiabilitat de la pista d'auditoria i la preservació de dades crítiques al llarg del cicle de vida. La documentació resultant (clarament traçable, defensable i proporcional al risc) constitueix una evidència important durant les auditories reguladores.
També és crucial establir una bona governança del sistema. Les agències hi estan d'acord No hi pot haver integritat de dades sense responsabilitats ben definides. S'han de distingir clarament rols com el propietari del sistema, el propietari del procés, la qualitat, la informàtica i l'automatització, assegurant que cada àrea entén i assumeix el seu paper. Aquesta estructura minimitza les ambigüitats, garanteix decisions ràpides i documentades i redueix els errors derivats de la falta de claredat en la gestió de canvis, configuracions o revisions periòdiques.
La cultura organitzativa representa un altre element crític. La FDA, l'EMA i la MHRA subratllen que cap tecnologia pot compensar una cultura feble o permissiva davant les males pràctiques. El formació contínua, rigorosa i adaptada al rol Assegura que el personal entén l'impacte de les seves decisions en la integritat de les dades. De la mateixa manera, la promoció d'un entorn on es fomenta la comunicació oberta, s'evita la pressió per assolir els resultats i es desaconsella pràctiques com el "backdating" o la "prova de compliment", sosté un sistema fiable a llarg termini.
Finalment, el revisió periòdica del rendiment del sistema, combinat amb auditories internes ben estructurades, confirma que tots els controls segueixen sent operatius i efectius. El seguiment continu de les pistes d'auditoria, la verificació regular d'accés i el seguiment d'incidències o desviacions són mecanismes principals per demostrar que el sistema continua funcionant dins d'un estat validat. La FDA i la MHRA subratllen la importància d'aplicar mesures correctives oportunes basades en una anàlisi de la causa arrel, assegurant que qualsevol desviació relacionada amb la integritat de les dades es gestiona amb el mateix rigor que una fallada del procés.
En general, la implementació d'un sistema robust no depèn només d'eines tecnològiques avançades, sinó de la integració coherent del disseny, validació, govern, formació i supervisió contínua. Aquest enfocament holístic és l'únic capaç de garantir un marc que sigui sostenible, defensable i totalment alineat amb les expectatives de la FDA, EMA i MHRA.
4. Casos típics d'incompliment de la integritat de les dades segons FDA, EMA i MHRA
S'han identificat inspeccions de les principals agències reguladores patrons recurrents d'incompliment pel que fa a la integritat de les dades. Aquests casos no només reflecteixen fallades tècniques, sinó també llacunes culturals, deficiències en el govern de les dades i la manca de controls adequats en els processos GxP. Conèixer aquests escenaris facilita que les organitzacions anticipin les observacions crítiques i enforteixin els seus sistemes abans d'una auditoria.
Manca de control sobre la pista d'auditoria
Un dels troballes més habituals a les cartes d'advertència i als informes de deficiències. Els problemes típics inclouen:
-
- Pista d'auditoria desactivada o limitada a canvis administratius.
- Registres incomplets (p. ex., només canvien els paràmetres, però no s'eliminen).
- Revisions inexistents o realitzades per personal no qualificat.
- Absència d'acreditació documental de la revisió.
- Agències més estrictes: MHRA i FDA, que consideren que la manca de revisió és un incompliment important.
Comptes compartits o "genèrics".
L'ús de comptes col·lectius ("administrador", "operador", "laboratori") impedeix l'atribució fiable de qui ha realitzat una acció. Aquesta fallada viola directament el principi atribuïble (A) d'ALCOA i normalment condueix a observacions crítiques sobre el control d'accés i la seguretat del sistema.
Conseqüència comuna: necessitat de revalidar el sistema i reciclar tot el personal.
Modificació o supressió de dades sense traçabilitat
Casos típics trobats en laboratori i fabricació:
-
- S'han eliminat cromatogrames abans de la revisió.
- Resultats repetits sense una justificació clara (“prova de compliment”).
- Retractament de dades sense registre del motiu.
- Canvis de paràmetres en PLC/SCADA sense documentació en Control de canvis.
La FDA considera aquest tipus de comportament falsificació de dades, encara que la intenció no fos maliciosa.
Registres manuals incomplets o reescrits
La manipulació de dades en paper continua sent una de les troballes més freqüents, fins i tot en plantes altament digitalitzades. Exemples comuns:
-
- Esborrats sense signatura i data.
- Dades afegides més tard sense aclariments ("backdating").
- Resultats transcrits incorrectament des d'ordinadors.
- No es permeten els formats duplicats per a "assaigs anteriors".
L'EMA destaca que els registres manuscrits han de ser contemporanis, complets i verificats.
Manca de validació dels sistemes informàtics crítics
Un sistema no validat es considera a font potencial de dades poc fiables. Descobriments típics:
-
- Absència d'URS clara.
- Registre insuficient de les proves realitzades.
- Manca de proves de proves de seguretat o còpies de seguretat.
- Versions de programari en ús que no corresponen a la versió validada.
- MHRA ha publicat diversos exemples on la manca de CSV provoca deficiències crítiques.
Drets d'accés inadequats
-
- Autoritzacions administratives concedides als operadors.
- Falta de segregació entre creació, revisió i aprovació.
- Perfils inexistents o poc definits.
- Això s'associa amb un alt risc de manipulació, accidental o intencionada.
Deficiències en còpia de seguretat i recuperació de dades
Els reguladors esperen còpies automatitzades, verificades i segures. Casos típics:
-
- Còpies de seguretat no provades.
- Còpies emmagatzemades en dispositius no protegits (USB, discos no controlats).
- Recuperació impossible o incompleta després d'un incident.
- Aquesta troballa afecta especialment els sistemes SCADA, els PLC i els servidors locals.
Manca de coherència entre els registres electrònics i les operacions reals
Molt observat en les inspeccions de la FDA i MHRA:
-
- Temps de procés que no coincideixen amb els registres.
- Accions de planta no reflectides a la pista d'auditoria.
- Equips que funcionen fora de les especificacions sense alarma ni registre.
Aquest tipus d'incoherències sovint condueixen a auditories ampliades i revisions retrospectives.
SOP obsolets o no aplicats
Fins i tot amb una tecnologia robusta, un error de procediment pot comprometre completament la integritat de les dades. Exemples:
-
- Procediments que no inclouen la revisió de la pista d'auditoria.
- Instruccions contradictòries entre departaments.
- Instruccions massa generals que permeten interpretacions diverses.
L'EMA destaca que la governança dels documents és un component bàsic de la integritat de les dades.
Reflexió final sobre el compliment de Data Integrity
Tot i que la FDA, l'EMA i la MHRA coincideixen en l'objectiu principal de protegir la integritat de les dades, cada autoritat introdueix matisos que afecten l'estratègia de compliment. La clau és desenvolupar un sistema alineat amb l'enfocament europeu (EMA/MHRA), que sigui el més complet i exigent, però prou flexible per satisfer els requisits operatius de la FDA. En resum, la integritat de les dades no es garanteix només mitjançant la tecnologia, sinó a través d'un forta cultura organitzativa, processos ben dissenyats i un marc de govern que garanteix que la informació crítica segueixi sent fiable durant tot el seu cicle de vida.