La Integridad de Datos (Data Integrity) es un pilar central en la industria farmacéutica y en cualquier entorno regulado bajo GxP. Tanto la FDA (Estados Unidos), como la EMA (Europa) y la MHRA (Reino Unido) han desarrollado guías específicas que abordan los requisitos para garantizar que los datos generados a lo largo del ciclo de vida del producto sean completos, consistentes, precisos, disponibles y atribuibles.
Aunque los principios fundamentales son compartidos, cada autoridad introduce matices importantes que condicionan la forma en que las empresas deben implementar sus sistemas de control, automatización y aseguramiento de la calidad.
Este artículo ofrece una comparativa práctica para facilitar a las organizaciones la aplicación de controles robustos y defendibles ante auditorías.
1. Principios fundamentales compartidos (FDA, EMA, MHRA)
Aunque cada agencia reguladora aporta matices propios, las tres coinciden en un conjunto de principios esenciales que conforman la base de cualquier sistema de Integridad de Datos sólido. Estos principios se aplican a todos los procesos GxP y abarcan desde la captura y el procesamiento inicial del dato hasta su archivo, acceso, revisión y recuperación durante todo el ciclo de vida.
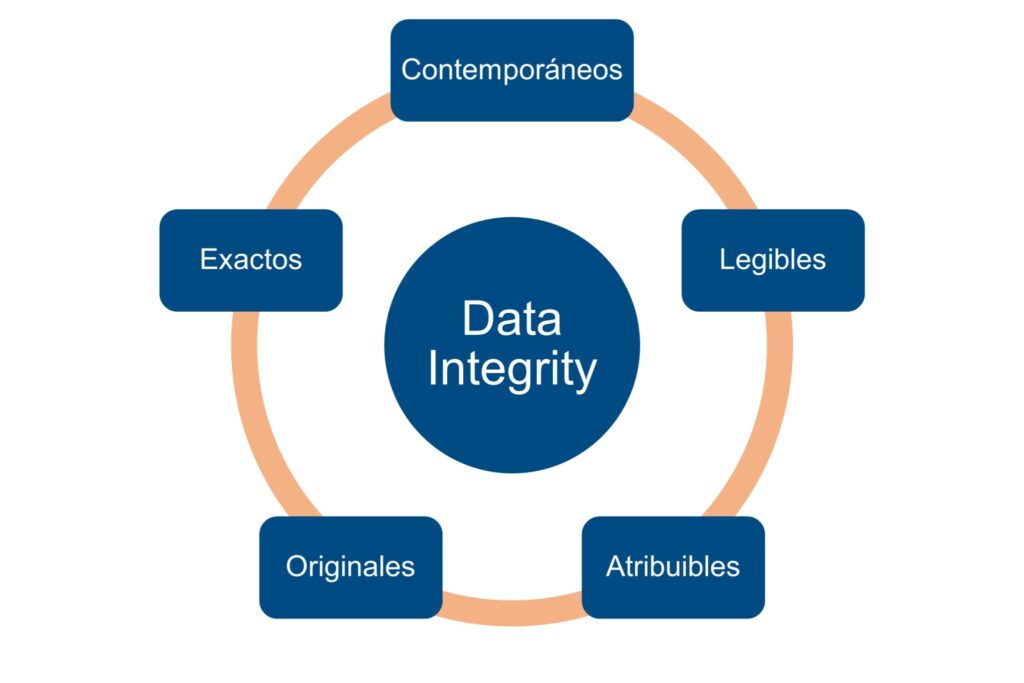
1.1. Integridad de Datos sustentada en ALCOA+
Las tres autoridades reguladoras consideran ALCOA+ como el estándar conceptual para definir qué es un dato confiable en contextos GxP. Los datos deben ser:
- Atribuibles: asociados claramente a una persona, equipo o sistema.
- Legibles: comprensibles durante todo el ciclo de vida.
- Contemporáneos: registrados en el momento en que ocurren.
- Originales: conservados en su forma primaria o con copias verdaderas.
- Exactos: libres de manipulación y errores.
Y, además: Completos, Consistentes, Duraderos y Disponibles.
La evolución regulatoria impulsada por la digitalización ha llevado incluso a la adopción del término ALCOA++en documentos recientes de la EMA, particularmente en las guías para sistemas computarizados y datos electrónicos en ensayos clínicos (GCP). Esta expansión busca incorporar requisitos aún más avanzados de trazabilidad y gobernanza de datos para manejar sistemas informáticos sofisticados y el uso de tecnologías híbridas.
Este marco conceptual es la base de cada inspección de FDA, EMA y MHRA, que ahora evalúan no solo el dato en sí, sino también la trazabilidad lógica entre registros, decisiones y evidencias.
1.2. Ciclo de vida del dato: enfoque holístico y continuo
Las tres agencias entienden la Integridad de Datos como un proceso transversal que se extiende más allá de la mera adquisición:
- Generación
- Procesamiento
- Revisión
- Reporte
- Archivado
- Recuperación
- Modificación (controlada)
- Retirada o destrucción
FDA, EMA y MHRA esperan que cada etapa cuente con controles técnicos y procedimentales, y que la organización pueda demostrar su robustez en auditorías.
1.3. Enfoque basado en riesgo
La gobernanza del dato aparece de forma explícita en MHRA y de manera transversal en FDA y EMA. Todas coinciden en que la Integridad de Datos no puede depender únicamente de herramientas tecnológicas. Debe existir:
- Cultura de transparencia.
- Roles y responsabilidades claros.
- Tolerancia cero a manipulaciones o retrasos en el registro.
- Disponibilidad de recursos suficientes en QA.
- Autonomía del área de calidad para revisar, bloquear o investigar registros.
Este enfoque cultural es ahora un criterio clave en inspecciones: los reguladores buscan demostrar si la empresa “vive” la integridad de datos o simplemente la documenta.
1.4. Enfoque basado en riesgo según ICH Q9
La gestión del riesgo regula cómo deben aplicarse los controles. FDA, EMA y MHRA esperan:
- Identificación de vulnerabilidades del dato.
- Clasificación del riesgo según impacto en producto, paciente e integridad.
- Mitigación alineada con GAMP 5 (2nd Ed.).
- Revisión continua del riesgo tras cambios en software, procesos o personal.
En auditorías, los inspectores ya no preguntan solo por SOPs: ahora piden evidencia de que los riesgos del dato han sido identificados, evaluados y mitigados de forma activa.
1.5. Controles técnicos robustos: Audit Trail, permisos y trazabilidad
Las tres agencias coinciden en que los sistemas informatizados deben integrar controles que prevengan la manipulación:
- Audit Trail seguro e inalterable, que registre quién hizo qué, cuándo y por qué.
- Gestión de accesos basada en roles, sin cuentas genéricas ni permisos innecesarios.
- Control de versiones, tanto de software como de configuraciones críticas.
- Respaldo y recuperación de datos, validado y probado regularmente.
- Sincronización horaria, fundamental para correlación de eventos.
La diferencia radica en el grado de exigencia (MHRA es la más estricta), pero el principio es idéntico.
1.6. Revisión periódica del audit trail y del dato crítico
Se trata de uno de los elementos de mayor atención en inspecciones durante los últimos años. FDA, EMA y MHRA exigen:
- Revisión rutinaria del audit trail basada en riesgos.
- Evidencia de que las revisiones son independientes, oportunas y competentes.
- Capacitación específica del personal revisor.
- Coherencia entre registro electrónico y operaciones reales.
- No basta con “activar” el audit trail: debe ser útil, revisado y defendible.
1.7. Validación del sistema informatizado (CSV/CSA) como parte básica del control
Los reguladores coinciden en que un sistema no validado es un sistema que no garantiza integridad de datos. Expectativas comunes:
- Validación proporcional al riesgo (GAMP 5 / CSA).
- Trazabilidad completa URS → Riesgos → Pruebas → Evidencias.
- Pruebas significativas y no redundantes.
- Gestión del ciclo de vida del sistema: cambios, incidencias, mantenimiento.
FDA ha impulsado la visión CSA, pero EMA y MHRA coinciden en su enfoque basado en riesgo.
2. Diferencias entre FDA, EMA y MHRA
A continuación, se recogen las principales diferencias entre la FDA, EMA y MHRA con respecto al enfoque y terminología, grado de independencia del audit trail, expectativas sobre sistemas híbridos y revisión de datos y segregación de responsabilidades:
Enfoque y terminología
| FDA | Adopta un tono más operativo, centrado en casos reales observados durante inspecciones. Su enfoque es pragmático desde el punto de vista del cumplimiento. Publicación clave: “Data Integrity and Compliance With Drug CGMP Questions and Answers”. |
| EMA | Integra Data Integrity dentro del marco de GMP europeo, vinculando la integridad de datos con el Sistema de Calidad Farmacéutico (ICH Q10). Se orienta a expectativas estratégicas: gobierno del sistema, roles y responsabilidades. |
| MHRA | Es la autoridad históricamente más estricta. Su guía va más allá de GMP e introduce conceptos como el «data governance system» y un elevado nivel de madurez en controles. |
Grado de independencia del Audit trail
| FDA | Recalca la necesidad de que el audit trail sea seguro y no editable, pero no exige una arquitectura específica. |
| EMA | Insiste en que el audit trail se revise rutinariamente como parte del flujo de trabajo de calidad. |
| MHRA | Es clara: audit trails no pueden estar deshabilitados, deben estar completamente integrados y protegidos de accesos administrativos indebidos. |
Expectativas sobre sistemas híbridos
| FDA | Los acepta, pero advierte de sus riesgos: duplicidad de fuentes, retrasos, manipulación y errores manuales. |
| EMA | Promueve la migración progresiva hacia sistemas integrados. |
| MHRA | Es más contundente: los sistemas híbridos deben considerarse una solución transitoria, no permanente. |
Revisión de datos y segregación de responsabilidades
| FDA | Los acepta, pero advierte de sus riesgos: duplicidad de fuentes, retrasos, manipulación y errores manuales. |
| EMA | Promueve la migración progresiva hacia sistemas integrados. |
| MHRA | Es más contundente: los sistemas híbridos deben considerarse una solución transitoria, no permanente. |
Comparativa práctica
|
SEGURIDAD DE SISTEMAS INFORMATIZADOS |
|||
|
Requisito |
FDA | EMA |
MHRA |
|
Gestión de accesos |
Necesaria, basada en roles | Integrada en PQS | Muy estricta y detallada |
|
Audit trail |
Obligatorio y revisable | Obligatorio con revisión periódica | No puede ser deshabilitado |
|
Control de versiones |
Recomendado | Requerido | Requerido y supervisado |
|
REGISTROS EN PAPEL Y SISTEMAS HÍBRIDOS |
|||
|
Aspecto |
FDA | EMA |
MHRA |
|
Aceptación |
Limitada, con riesgos | Aceptados pero no preferidos | Transitorios, no recomendados |
|
Principales riesgos |
Errores, duplicidad | Falta de trazabilidad | Manipulación y pérdida de control |
|
DATA GOVERNANCE |
|||
| Elemento | FDA | EMA |
MHRA |
|
Modelo formal de «gobernanza del dato» |
No explícito | Integrado al PQS | Requisito explícito y detallado |
3. Cómo implementar un sistema robusto que cumpla con FDA, EMA y MHRA
Implementar un sistema de Integridad de Datos verdaderamente robusto implica ir más allá del cumplimiento documental y avanzar hacia un marco integral que combine tecnología, cultura organizativa, gobernanza clara y supervisión continua. Aunque cada agencia tiene particularidades, todas coinciden en que la Integridad de Datos debe gestionarse a lo largo de todo el ciclo de vida, desde el diseño del sistema hasta su retirada. Esto significa que los requisitos no pueden abordarse de forma reactiva; deben integrarse desde etapas tempranas para asegurar que el sistema sea inherentemente confiable, defendible y capaz de resistir auditorías rigurosas.
Un primer paso crítico consiste en adoptar un enfoque “Data Integrity by Design”. Este principio, cada vez más presente en las guías de FDA y EMA, implica configurar sistemas digitalizados y equipos GxP con capas de control técnico pensadas desde su concepción. La correcta definición de requisitos de usuario (URS) es fundamental para asegurar que las funcionalidades principales estén incorporadas antes de la validación, como trazabilidad, audit trail inalterable, gestión de accesos o controles de versión. Un sistema incapaz de registrar datos de forma segura o que no permita restringir adecuadamente los permisos de usuario será, por definición, un sistema no conforme, por mucho que se complemente con procedimientos.
La validación del sistema informatizado (CSV), alineada con GAMP 5 y las expectativas CSA de FDA, es otro pilar fundamental. La validación debe demostrar que el sistema funciona tal y como se diseñó, que protege los datos mediante controles técnicos robustos y que opera en estado controlado bajo condiciones rutinarias. Este proceso debe incluir pruebas específicas que aseguren la integridad de los registros electrónicos, la confiabilidad del audit trail y la preservación de datos críticos durante todo el ciclo de vida. La documentación resultante (claramente trazable, defendible y proporcional al riesgo) constituye una evidencia importante durante auditorías regulatorias.
El establecimiento de una gobernanza sólida del sistema es igualmente determinante. Las agencias coinciden en que no puede existir Integridad de Datos sin responsabilidades bien definidas. Deben distinguirse claramente roles como propietario del sistema, propietario del proceso, calidad, TI y automatización, asegurando que cada área entiende y asume su papel. Esta estructura minimiza ambigüedades, garantiza decisiones rápidas y documentadas, y reduce fallos derivados de la falta de claridad en la gestión de cambios, configuraciones o revisiones periódicas.
La cultura organizativa representa otro elemento crítico. FDA, EMA y MHRA enfatizan que ninguna tecnología puede compensar una cultura débil o permisiva frente a malas prácticas. La formación continua, rigurosa y adaptada al rol asegura que el personal comprende el impacto de sus decisiones en la integridad del dato. Del mismo modo, promover un ambiente donde se fomente la comunicación abierta, se evite la presión por cumplir resultados y se desalienten prácticas como “backdating” o “testing into compliance” sostiene un sistema confiable a largo plazo.
Finalmente, la revisión periódica del desempeño del sistema, combinada con auditorías internas bien estructuradas, permite confirmar que todos los controles permanecen operativos y eficaces. La supervisión continua de audit trails, la verificación regular de accesos y la monitorización de incidentes o desviaciones constituyen mecanismos principales para demostrar que el sistema sigue funcionando dentro de un estado validado. FDA y MHRA recalcan la importancia de aplicar medidas correctivas oportunas y basadas en un análisis de causa raíz, asegurando que cualquier desviación relacionada con Integridad de Datos se gestiona con la misma rigurosidad que un fallo de proceso.
En conjunto, la implementación de un sistema robusto no depende únicamente de herramientas tecnológicas avanzadas, sino de la integración coherente de diseño, validación, gobernanza, formación y supervisión continua. Este enfoque holístico es el único capaz de garantizar un marco sostenible, defendible y plenamente alineado con las expectativas de FDA, EMA y MHRA.
4. Casos típicos de incumplimiento de Data Integrity según FDA, EMA y MHRA
Las inspecciones de las principales agencias reguladoras han identificado patrones recurrentes de incumplimiento en materia de Integridad de Datos. Estos casos no solo reflejan fallos técnicos, sino también brechas culturales, deficiencias en la gobernanza del dato y ausencia de controles adecuados en los procesos GxP. Conocer estos escenarios facilita a las organizaciones anticiparse a observaciones críticas y fortalecer sus sistemas antes de una auditoría.
Falta de control sobre el Audit Trail
Uno de los hallazgos más habituales en Warning Letters y Deficiency Reports. Problemas típicos incluyen:
-
- Audit trail desactivado o limitado a cambios administrativos.
- Registros incompletos (p. ej., únicamente cambios de parámetros, pero no borrados).
- Revisiones inexistentes o realizadas por personal no cualificado.
- Ausencia de evidencia documental de la revisión.
- Agencias más estrictas: MHRA y FDA, que consideran la falta de revisión un incumplimiento mayor.
Cuentas compartidas o “genéricas”
El uso de cuentas colectivas (“admin”, “operator”, “lab”) impide atribuir de forma confiable quién realizó una acción. Este fallo vulnera directamente el principio de Attributable (A) de ALCOA y suele derivar en observaciones críticas sobre control de accesos y seguridad del sistema.
Consecuencia habitual: necesidad de revalidar el sistema y reentrenar a todo el personal.
Modificación o eliminación de datos sin trazabilidad
Casos típicos encontrados en laboratorio y fabricación:
-
- Cromatogramas eliminados antes de la revisión.
- Resultados repetidos sin justificación clara (“testing into compliance”).
- Reprocesamiento de datos sin registro del motivo.
- Cambios de parámetros en PLC/SCADA sin documentación en Change Control.
FDA considera este tipo de comportamiento como falsificación de datos, incluso si la intención no fue maliciosa.
Registros manuales incompletos o reescritos
La manipulación de datos en papel sigue siendo uno de los hallazgos más frecuentes, incluso en plantas altamente digitalizadas. Ejemplos comunes:
-
- Tachaduras sin firma y fecha.
- Datos añadidos posteriormente sin aclaración (“backdating”).
- Resultados transcritos incorrectamente desde equipos.
- Formatos duplicados para “ensayos previos” no permitidos.
EMA subraya que los registros manuscritos deben ser contemporáneos, completos y verificados.
Falta de validación de sistemas informatizados críticos
Un sistema no validado se considera una fuente potencial de datos no confiables. Hallazgos típicos:
-
- Ausencia de una URS clara.
- Registro insuficiente de pruebas realizadas.
- Falta de evidencia de pruebas de seguridad o backup.
- Versiones de software en uso que no corresponden con la versión validada.
- MHRA ha publicado múltiples ejemplos donde la falta de CSV deriva en deficiencias críticas.
Derechos de acceso inadecuados
-
- Permisos administrativos otorgados a operadores.
- Falta de segregación entre creación, revisión y aprobación.
- Perfiles inexistentes o mal definidos.
- Esto se asocia con un riesgo elevado de manipulación, accidental o intencionada.
Deficiencias en Backup y Recuperación de datos
Los reguladores esperan copias automáticas, verificadas y seguras. Casos típicos:
-
- Backups no probados.
- Copias almacenadas en dispositivos no protegidos (USB, discos no controlados).
- Recuperación imposible o incompleta tras un incidente.
- Este hallazgo afecta especialmente a sistemas SCADA, PLC y servidores locales.
Falta de coherencia entre los registros electrónicos y las operaciones reales
Muy observado en inspecciones FDA y MHRA:
-
- Tiempos de proceso que no coinciden con los registros.
- Acciones en planta no reflejadas en el audit trail.
- Equipos funcionando fuera de especificación sin alarma o registro.
Este tipo de incoherencias suele derivar en auditorías ampliadas y revisiones retrospectivas.
SOPs desactualizados o no aplicados
Incluso con tecnología robusta, un fallo en los procedimientos puede comprometer por completo la integridad del dato. Ejemplos:
-
- Procedimientos que no contemplan la revisión del audit trail.
- Instrucciones contradictorias entre departamentos.
- Instrucciones demasiado generales que permiten interpretaciones diversas.
EMA enfatiza que la gobernanza documental es un componente básico de la Integridad de Datos.
Reflexión final sobre el cumplimiento en Integridad de Datos
Aunque FDA, EMA y MHRA coinciden en el principal propósito de proteger la integridad del dato, cada autoridad introduce matices que afectan la estrategia de cumplimiento. La clave es desarrollar un sistema alineado con el enfoque europeo (EMA/MHRA), que es el más completo y exigente, pero suficientemente flexible para satisfacer los requisitos operativos de la FDA. En definitiva, la integridad del dato no se garantiza solo mediante tecnología, sino mediante una cultura organizativa sólida, procesos bien diseñados y un marco de gobernanza que asegure que la información crítica permanece confiable durante todo su ciclo de vida.