The Data Integrity It is a central pillar in the pharmaceutical industry and in any environment regulated under GxP. Both the FDA (United States), like the EMA (Europe) and the MHRA (United Kingdom) have developed specific guides that address requirements to ensure that data generated throughout the product lifecycle is complete, consistent, accurate, available and attributable.
Although the fundamental principles are shared, each authority introduces important nuances that determine the way in which companies must implement their control, automation and quality assurance systems.
This article offers a practical comparison to make it easier for organizations to apply robust and audit-defensible controls.
1. Shared fundamental principles (FDA, EMA, MHRA)
Although each regulatory agency provides its own nuances, All three agree on a set of essential principles that form the basis of any solid Data Integrity system. These principles apply to all GxP processes and range from initial data capture and processing to archiving, access, review and retrieval throughout the lifecycle.
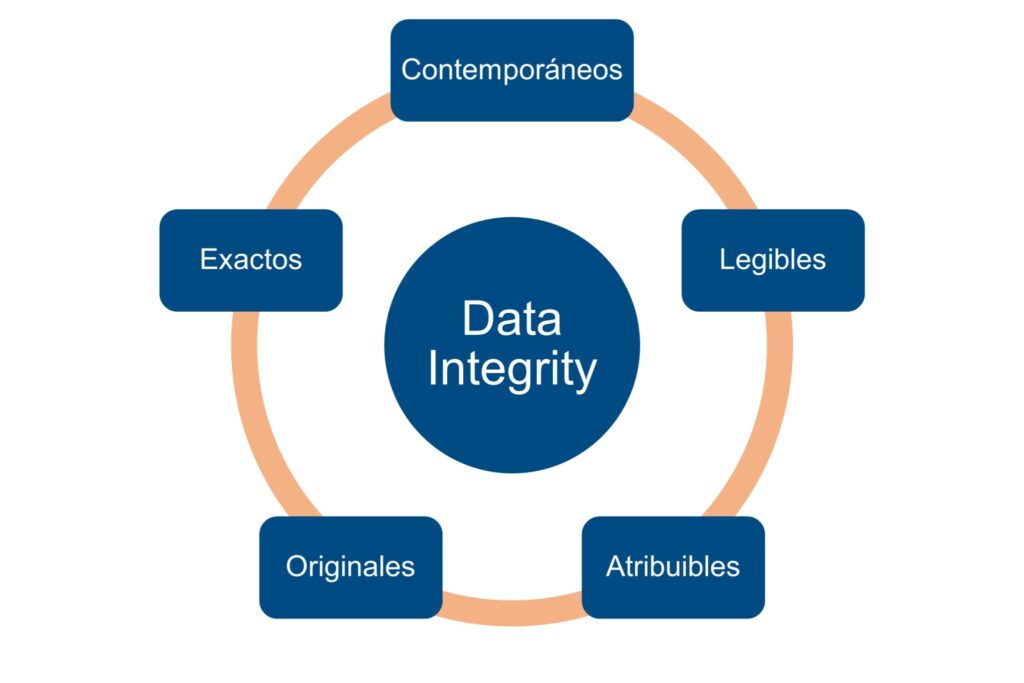
1.1. Data Integrity supported by ALCOA+
The three regulatory authorities consider ALCOA+ as the conceptual standard to define what is reliable data in GxP contexts. The data must be:
- Attributable: clearly associated with a person, team or system.
- Legible: understandable throughout the life cycle.
- Contemporaries: recorded at the time they occur.
- Originals: preserved in their primary form or with true copies.
- Exact: free from manipulation and errors.
And, furthermore: Complete, Consistent, Durable and Available.
Regulatory evolution driven by digitalization has even led to the adoption of the term ALCOA++ in recent EMA documents, particularly in the guidelines for computerized systems and electronic data in clinical trials (GCP). This expansion seeks to incorporate even more advanced data traceability and governance requirements to handle sophisticated IT systems and the use of hybrid technologies.
This conceptual framework is the basis of each FDA, EMA and MHRA inspection, which now evaluate not only the data itself, but also the logical traceability between records, decisions and evidence.
1.2. Data life cycle: holistic and continuous approach
The three agencies understand Data Integrity as a transversal process that extends beyond mere acquisition:
- Generation
- Prosecution
- Revision
- Report
- Filed
- Recovery
- Modification (controlled)
- Withdrawal or destruction
FDA, EMA and MHRA expect each stage to have technical and procedural controls, and that the organization can demonstrate its robustness in audits.
1.3. Risk-based approach
Data governance appears explicitly in MHRA and transversally in FDA and EMA. They all agree that the Data Integrity cannot depend solely on technological tools. There must be:
- Culture of transparency.
- Clear roles and responsibilities.
- Zero tolerance for manipulations or delays in registration.
- Availability of sufficient resources in QA.
- Autonomy of the quality area to review, block or investigate records.
This cultural approach is now a key criterion in inspections: regulators seek to demonstrate whether the company “lives” data integrity or simply documents it.
1.4. Risk-based approach according to ICH Q9
The Risk management regulates how controls should be applied. FDA, EMA and MHRA expect:
- Identification of data vulnerabilities.
- Risk classification according to impact on product, patient and integrity.
- Mitigation aligned with GAMP 5 (2nd Ed.).
- Continuous risk review after changes in software, processes or personnel.
In audits, inspectors no longer just ask about SOPs: they now ask for evidence that data risks have been identified, evaluated and actively mitigated.
1.5. Robust technical controls: Audit Trail, permits and traceability
The three agencies agree that computerized systems must integrate controls that prevent manipulation:
- Secure and unalterable Audit Trail, recording who did what, when and why.
- Role-based access management, without generic accounts or unnecessary permissions.
- Version control, both of software and critical configurations.
- Data backup and recovery, regularly validated and tested.
- Time synchronization, essential for correlation of events.
The difference lies in the degree of requirement (MHRA is the strictest), but the principle is identical.
1.6. Periodic review of the audit trail and critical data
It is one of the elements of greater attention in inspections during the last few years. FDA, EMA and MHRA require:
- Routine risk-based audit trail review.
- Evidence that the reviews are independent, timely and competent.
- Specific training of reviewing staff.
- Coherence between electronic registration and real operations.
- It is not enough to “activate” the audit trail: it must be useful, reviewed and defensible.
1.7. Validation of the computerized system (CSV/CSA) as a basic part of control
Regulators agree that a non-validated system is a system that does not guarantee data integrity. Common expectations:
- Validation proportional to risk (GAMP 5 / CSA).
- Complete traceability URS → Risks → Tests → Evidence.
- Meaningful and non-redundant tests.
- System life cycle management: changes, incidents, maintenance.
FDA has pushed the CSA view, but EMA and MHRA agree on their risk-based approach.
2. Differences between FDA, EMA and MHRA
Below are the main differences between the FDA, EMA and MHRA with respect to the approach and terminology, degree of independence of the audit trail, expectations for hybrid systems and data review and segregation of responsibilities:
Approach and terminology
| FDA | It adopts a more operational tone, focusing on real cases observed during inspections. Their approach is pragmatic from a compliance point of view. Key post: “Data Integrity and Compliance With Drug CGMP Questions and Answers”. |
| EMA | Integrates Data Integrity within the European GMP framework, linking data integrity with the Pharmaceutical Quality System (ICH Q10). It is oriented towards strategic expectations: system governance, roles and responsibilities. |
| MHRA | It is the historically strictest authority. Its guide goes beyond GMP and introduces concepts such as the “data governance system” and a high level of maturity in controls. |
Degree of independence of the Audit trail
| FDA | It emphasizes the need for the audit trail to be secure and non-editable, but does not require a specific architecture. |
| EMA | Insist that the audit trail be routinely reviewed as part of the quality workflow. |
| MHRA | It is clear: audit trails cannot be disabled, they must be fully integrated and protected from improper administrative access. |
Expectations about hybrid systems
| FDA | He accepts them, but warns of their risks: duplication of sources, delays, manipulation and manual errors. |
| EMA | Promotes progressive migration towards integrated systems. |
| MHRA | It is more forceful: hybrid systems should be considered a temporary solution, not a permanent one. |
Data review and segregation of responsibilities
| FDA | He accepts them, but warns of their risks: duplication of sources, delays, manipulation and manual errors. |
| EMA | Promotes progressive migration towards integrated systems. |
| MHRA | It is more forceful: hybrid systems should be considered a temporary solution, not a permanent one. |
Practical comparison
|
COMPUTER SYSTEMS SECURITY |
|||
|
Requirement |
FDA | EMA |
MHRA |
|
Access management |
Necessary, role-based | Integrated in PQS | Very strict and detailed |
|
Audit trail |
Mandatory and reviewable | Mandatory with periodic review | Cannot be disabled |
|
Version control |
Recommended | Requested | Required and supervised |
|
PAPER RECORDS AND HYBRID SYSTEMS |
|||
|
Aspect |
FDA | EMA |
MHRA |
|
Acceptance |
Limited, with risks | Accepted but not preferred | Transient, not recommended |
|
Main risks |
Errors, duplicity | Lack of traceability | Manipulation and loss of control |
|
DATA GOVERNANCE |
|||
| Element | FDA | EMA |
MHRA |
|
Formal “data governance” model |
Not explicit | Integrated into PQS | Explicit and detailed requirement |
3. How to implement a robust system that complies with FDA, EMA and MHRA
Implementing a truly robust Data Integrity system involves going beyond documentary compliance and moving towards a Comprehensive framework that combines technology, organizational culture, clear governance and continuous supervision. Although each agency has particularities, they all agree that Data Integrity must managed throughout the entire life cycle, from system design to retirement. This means that requirements cannot be addressed reactively; They must be integrated from early stages to ensure that the system is inherently reliable, defensible, and able to withstand rigorous audits.
A critical first step is to adopt a “Data Integrity by Design” approach. This principle, increasingly present in FDA and EMA guidelines, involves configuring digitalized systems and GxP equipment with layers of technical control designed from their conception. The correct definition of user requirements (URS) is essential to ensure that the main functionalities are incorporated before validation, such as traceability, unalterable audit trail, access management or version controls. A system that is unable to securely record data or that does not allow user permissions to be appropriately restricted will, by definition, be a non-compliant system, no matter how supplemented it is with procedures.
Computerized system validation (CSV), aligned with GAMP 5 and FDA CSA expectations, is another fundamental pillar. Validation must demonstrate that the system functions as designed, that it protects data through robust technical controls, and that it operates in a controlled state under routine conditions. This process must include specific tests that ensure the integrity of electronic records, the reliability of the audit trail and the preservation of critical data throughout the life cycle. The resulting documentation (clearly traceable, defensible and proportional to risk) constitutes important evidence during regulatory audits.
Establishing sound governance of the system is equally crucial. The agencies agree that There can be no Data Integrity without well-defined responsibilities. Roles such as system owner, process owner, quality, IT and automation must be clearly distinguished, ensuring that each area understands and assumes its role. This structure minimizes ambiguities, guarantees quick and documented decisions, and reduces failures derived from lack of clarity in the management of changes, configurations or periodic reviews.
Organizational culture represents another critical element. FDA, EMA and MHRA emphasize that no technology can compensate for a weak or permissive culture in the face of bad practices. The continuous, rigorous training adapted to the role Ensures that staff understand the impact of their decisions on data integrity. Likewise, promoting an environment where open communication is encouraged, pressure to meet results is avoided, and practices such as “backdating” or “testing into compliance” are discouraged, sustains a reliable system in the long term.
Finally, the periodic review of system performance, combined with well-structured internal audits, confirms that all controls remain operational and effective. Continuous monitoring of audit trails, regular access verification, and monitoring of incidents or deviations are primary mechanisms to demonstrate that the system continues to operate within a validated state. FDA and MHRA emphasize the importance of applying timely corrective measures based on a root cause analysis, ensuring that any deviation related to Data Integrity is managed with the same rigor as a process failure.
Overall, the implementation of a robust system does not depend only on advanced technological tools, but on the coherent design integration, validation, governance, training and continuous supervision. This holistic approach is the only one capable of guaranteeing a framework that is sustainable, defensible and fully aligned with the expectations of the FDA, EMA and MHRA.
4. Typical cases of Data Integrity non-compliance according to FDA, EMA and MHRA
Inspections by major regulatory agencies have identified recurring patterns of non-compliance regarding Data Integrity. These cases not only reflect technical failures, but also cultural gaps, deficiencies in data governance and lack of adequate controls in GxP processes. Knowing these scenarios makes it easier for organizations to anticipate critical observations and strengthen their systems before an audit.
Lack of control over the Audit Trail
One of the most common findings in Warning Letters and Deficiency Reports. Typical problems include:
-
- Audit trail disabled or limited to administrative changes.
- Incomplete logs (e.g. only parameter changes, but not deleted).
- Non-existent revisions or carried out by unqualified personnel.
- Absence of documentary evidence of the review.
- Stricter agencies: MHRA and FDA, which consider lack of review a major non-compliance.
Shared or “generic” accounts
The use of collective accounts (“admin”, “operator”, “lab”) prevents reliable attribution of who performed an action. This failure directly violates ALCOA's Attributable (A) principle and usually leads to critical observations regarding access control and system security.
Common consequence: need to revalidate the system and retrain all staff.
Modification or deletion of data without traceability
Typical cases found in laboratory and manufacturing:
-
- Chromatograms removed before review.
- Repeated results without clear justification (“testing into compliance”).
- Reprocessing of data without registration of the reason.
- Parameter changes in PLC/SCADA without documentation in Change Control.
FDA considers this type of behavior data falsification, even if the intent was not malicious.
Incomplete or rewritten manual records
The data manipulation on paper continues to be one of the most frequent finds, even in highly digitalized plants. Common examples:
-
- Erasures without signature and date.
- Data added later without clarification (“backdating”).
- Results incorrectly transcribed from computers.
- Duplicate formats for “previous essays” not permitted.
EMA emphasizes that handwritten records must be contemporary, complete and verified.
Lack of validation of critical computerized systems
An unvalidated system is considered a potential source of untrustworthy data. Typical findings:
-
- Absence of a clear URS.
- Insufficient registration of tests carried out.
- Lack of evidence of security testing or backup.
- Software versions in use that do not correspond to the validated version.
- MHRA has published multiple examples where the lack of CSV results in critical deficiencies.
Inadequate access rights
-
- Administrative permits granted to operators.
- Lack of segregation between creation, review and approval.
- Non-existent or poorly defined profiles.
- This is associated with a high risk of tampering, accidental or intentional.
Deficiencies in Backup and Data Recovery
Regulators expect automated, verified and secure copies. Typical cases:
-
- Untested backups.
- Copies stored on unprotected devices (USB, uncontrolled disks).
- Impossible or incomplete recovery after an incident.
- This finding especially affects SCADA systems, PLCs and local servers.
Lack of consistency between electronic records and actual operations
Highly observed in FDA and MHRA inspections:
-
- Process times that do not match the records.
- Plant actions not reflected in the audit trail.
- Equipment operating out of specification without alarm or registration.
These types of inconsistencies often lead to expanded audits and retrospective reviews.
Outdated or unenforced SOPs
Even with robust technology, a procedural failure can completely compromise data integrity. Examples:
-
- Procedures that do not include the review of the audit trail.
- Contradictory instructions between departments.
- Too general instructions that allow for diverse interpretations.
EMA emphasizes that document governance is a basic component of Data Integrity.
Final reflection on Data Integrity compliance
Although FDA, EMA and MHRA agree on the main purpose of protecting data integrity, each authority introduces nuances that affect the compliance strategy. The key is to develop a system aligned with the European approach (EMA/MHRA), which is the most comprehensive and demanding, but flexible enough to satisfy the FDA's operational requirements. In short, data integrity is not guaranteed only through technology, but through a strong organizational culture, well-designed processes and a governance framework that ensures that critical information remains reliable throughout its lifecycle.